 Our projects
Our projects
The directions we are taking
The directions we are taking
Alterations of protein-RNA interaction can lead to a variety of diseases, including cancer. For instance, the aberrant expression of RBPs has proven to have an oncogenic effect. Gaining functional insights on the mechanisms driving the oncogenic effect of aberrant AS regulation will provide novel potential therapeutic targets. For this reason, we are interested in understanding the transcriptional regulation of splicing factors in cancer.
In this context we showed that FOXA1 functions as the primary orchestrator of alternative splicing regulation in prostate cancer as compared to other key oncogenes as AR, ERG, and MYC. We demonstrate that FOXA1 binds to the regulatory regions of splicing-related genes, including HNRNPK and SRSF1. By controlling trans-acting factor expression, FOXA1 calibrates alternative splicing toward dominant mRNA isoform production. This regulation impacts on the inclusion of ultra-conserved exons, known as poison exons triggering nonsense-mediated decay (NMD). Inclusion of the NMD-determinant FLNA exon 30 by FOXA1-controlled SRSF1 promotes cell growth in vitro and predicts disease recurrence.
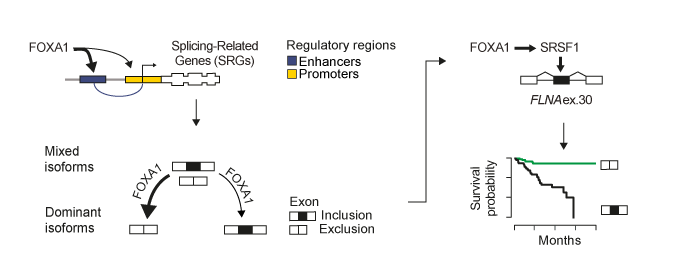
FOXA1 regulates alternative splicing in prostate cancer. FOXA1 is a master transcriptional regulator of splicing factors in prostate cancer. FOXA1 drives splice isoform production toward an optimal dominant mRNA product. FOXA1 controls exons triggering NMD, influencing prostate cancer patient prognosis. FOXA1-controlled SRSF1 enhances inclusion of FLNA exon 30, promoting disease recurrence
Overall, we show a new role for the pionner transcription factor FOXA1 in rewiring the alternative splicing landscape in prostate cancer through a cascade of events from chromatin access, to splicing factor regulation, and, finally, to alternative splicing of exons influencing patient survival. Our results are available onCell Reports!
RNA-binding proteins (RBPs) regulate splicing according to position-dependent principles, which can be exploited for analysis of regulatory motifs. In collaboration with the laboratory of Prof Jernej Ule, we developed RNAmotifs, a method that evaluates the sequence around differentially regulated alternative exons to identify clusters of short and degenerate sequences, referred to as multivalent RNA motifs. We showed that diverse RBPs share basic positional principles, but differ in their propensity to enhance or repress exon inclusion. We now are interested in understanding how these principles work in cancer tissues and developing novel methods to disantagle such complexity.
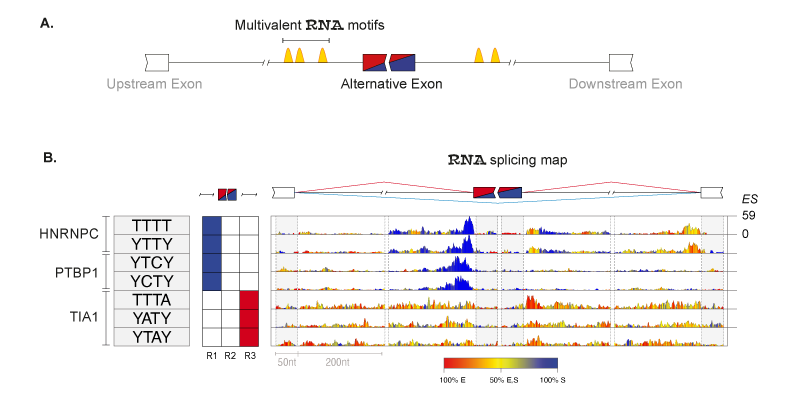
Principles of RNAmotifs algorithm. (A) Schematic rapresentation of multivalent motifs (i.e. cluster of short degenerate motifs) surrounding the alternative spliced exon. (B) RNA splicing maps of multivalent RNA motifs enriched in the 'mixed' set of exons regulated by hnRNP C, PTBP1 and TIA. Sequences of the enriched tetramers are shown on the left, followed by a color-coded panel showing the regions where tetramer enrichment reached the defined threshold around silenced (blue) or enhanced (red) exons. The right panel depicts the nucleotide-resolution RNA splicing map of each motif at the enhanced or silenced exons, and their flanking exons. The color key indicates whether the position-specific contribution originates from enhanced (red),silenced (blue),or both (yellow) sets. The maximum enrichment score (ES) value of the top tetramer is reported on the right.
Cancer is a disease dominated by heterogeneity whose effects are evident in the evolution and treatment of the disease. In the laboratory of Prof Francesca Ciccarelli, I characterized the genetics of synchronous colorectal cancers and demonstrated that tumors of the same patient are genetically heterogeneous with clear therapeutic implications. Characterizing the acquisition of clonal and subclonal aberrations is important to understand cancer progression and guide personalized medicine. To expand the number of the "actionable" targets, relying only on the mutational load of tumors is not sufficient (Bailey, Cell, 2018). To face this issue, we are currently studying somatic splicing aberration to uncover novel potential therapeutic targets.
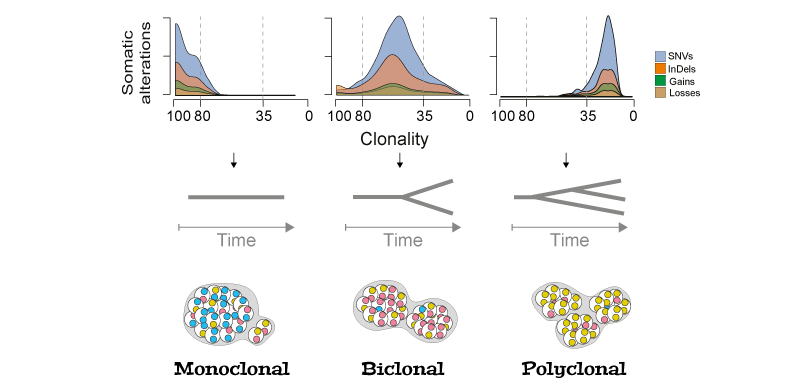
Tumor evolution reconstrunction. Schematic rapresentation of tumor clonal evolution reconstrunction. The density distributions of clonality for each type of alteration recapitulate the tumour clone composition (monoclonal, if the majority of alterations has clonality >80%; biclonal, if alterations accumulate between 35 and 80% clonality; or polyclonal if they have <35% clonality).
Details on clonal evolution of synchronous colorectal cancers are on Nature Communications!
We are currently leading the genomics analyses of Genomic Profiles Analysis in Children, Adolescents and Young Adult With Sarcomas (NCT04621201) trials
With the growth of high-throughput sequencing projects modern biology is facing novel bottlenecks due to Big Data issues. One of the challenges is to extract relevant information from this high-volume data while accounting for their intrinsic heterogeneity. So far, genomic screenings have profiled thousands of samples providing insights into the transcriptome of the cell. However, disentangling the heterogeneity of these transcriptomic Big Data to identify defective biological processes remains challenging. We addressed this challenge and introduced the novel concept of discretization of gene expression levels, which we derived from probabilistic modelling and shaped upon knowledge of RNA biology. Our Gene Set Enrichment Class Analysis (GSECA) algorithm exploits the bimodal behavior of RNA-sequencing gene expression profiles to identify altered gene sets in heterogeneous patient cohorts.
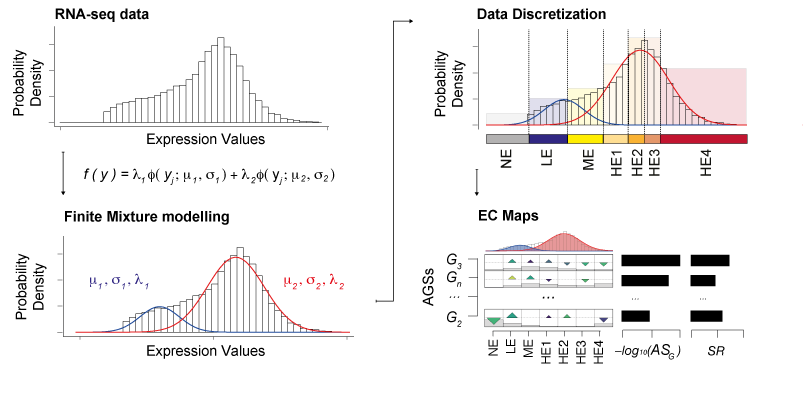
Schematic representation of GSECA algorithm. The algorithm proceeds through three sequential steps: (i) the sample-specific finite mixture modelling of gene expression distribution; (ii) the sample-specific discretization of expression values into seven categorical expression classes; and (iii) the statistical identification of altered gene sets (AGSs). The AGSs are visualized as expression class (EC) maps.
We showed that GSECA outperfomed 'state-of-art' algorithms in handling gene sets characterized by expression changes of groups of genes that are more intensively activated or repressed in a heterogeneous manner across samples. It can detect functionally related altered cell mechanisms in a condition of interest considering more heterogeneous cohorts as compared to other available methods. By boosting signal-to-noise ratio, GSECA can successfully manage the heterogeneity of thousands of samples and provides useful insights on clinical and biological patterns proper of a phenotype.
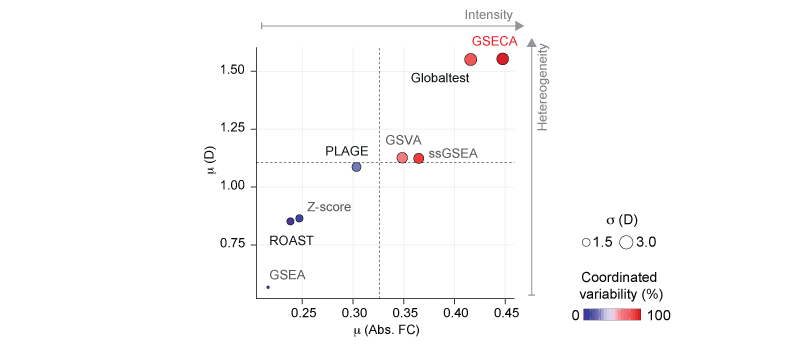
Performance evaluation of gene set analysis algorithms. Scatter plot of the mean absolute FC (Abs. FC), and dispersion (D) averaged on the gene sets detected by each method. Dot size represents the average standard deviation of D. Color key depicts the percentage of gene sets that contain both activated and repressed gene sets, namely coordinated variability.
By boosting signal-to-noise ratio, GSECA can successfully manage the heterogeneity of thousands of samples and provides useful insights on clinical and biological patterns proper of a phenotype. With this work we introduced the paradigm shift of "less is more" in treating large heterogenous RNA-seq datasets showing that it improves the detection of the altered biological processes in the phenotype of interest. Like looking to a bunch of photographs from a distance you might be able to get the big message!

GSECA metaphor. Heterogeneity can reveal a message if we look at it from a distance!
GSECA is out on Nucleic Acid Research!
In the N.A.R. paper, we generated a comprehensive assessment of the effect of PTEN loss across different cancer types. Our data showed that the impact of PTEN silencing on cellular program regulation is proportional to the impaired modulation of the PI3K/AKT signaling cascade, with the stronger effect of gliomas, endometrial, head and neck, breast carcinomas, melanomas, and sarcomas. GSECA correctly highlighted the role of PTEN in controlling immune-related processes in the majority of cancer types, particularly in those showing a significant alteration of the tumor immune-microenvironment (TIME) composition. These data support the importance of PTEN in modulating the immune system and therapy resistance.
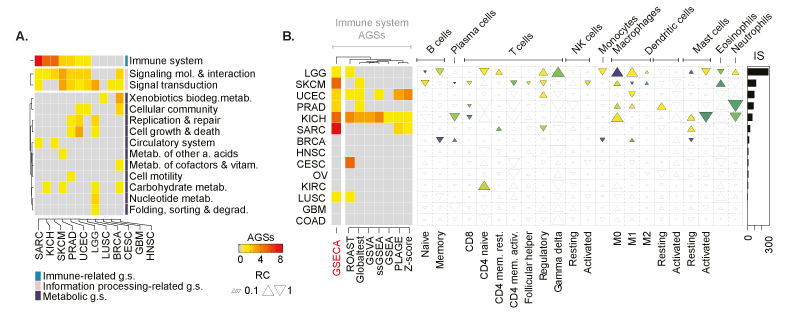
Pan-cancer analysis of PTEN somatic loss. . (A) Heatmap showing the altered classes of gene sets across cancer types. Classes are defined accordingly to the KEGG category. Each cell reports the number of AGSs. The annotation heatmap indicates the KEGG superclass of biological processes. (B) Heatmap shows the number of immune-related gene sets that are altered upon the loss of PTEN across cancer types accordingly to GSECA and the other GSA methods. On the right panel, EC map-like heatmap depicts the statistically significant alteration of the immune cell population across cancer types. The size of triangles the relative change of the percentage of tumor immune infiltrates between PTEN-loss and wild-type samples. The bar plot reports the IS for each cancer type.
Emerging evidence has suggested that PTEN loss is an immunosuppressive event in prostate tumors. However, the connection between PTEN and the immune system is complex and involves both pro- and anti-tumorigenic immune responses depending on the cellular phenotype and the TIME. Our analyes supported the notion that PTEN loss prostate cancers are non-T cell inflamed, or "cold", tumors. Futhermore, we showed that the immunosuppressive TIME of PTEN-loss prostate tumors could be driven by the significant activation of STAT3. PTEN loss were pivotal to show the shorter of disease-free survival of these patients and to underline the biomarker potential of PTEN expression levels.
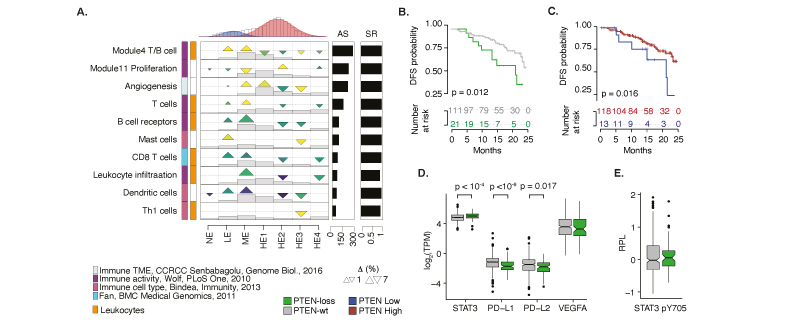
Impact of PTEN loss in prostate cancer. (A) GSECA EC map showing the altered immune expression signatures as a consequence of the somatic loss of PTEN in prostate cancer. (B). Disease-free survival (DFS) Kaplan-Mayer curves for PTEN-loss and PTEN-wt patients. (C). DSF Kaplan-Mayer curves measured stratifying PRAD patients on the optimal PTEN expression level (i.e. TPM=3.56, maximally selected rank statistics = 2.34) within two years from the initial treatment. (D) Boxplots showing expression distributions of PTEN normalized expression levels for PTEN-loss and PTEN-wt samples of four immune-response related genes. (E) Boxplot distributions of the relative level of STAT3 phosphorylation for PTEN-loss and PTEN-wt PRAD samples.
Details on our pancancer analysis of PTEN loss are on Nucleic Acid Research!
Artificial intelligence, or the discipline of developing computational algorithms able to perform tasks that requires human intelligence, is becoming a fundamental asset for healthcare and life science research. AI is the pivotal tool to exploit the information available in genomic Big Data and ultimately “deliver” a medicine of precision. The COVID-19 pandemic has opened up new possibilities for AI development. From the first drafts of the human genome, 20 years ago, the number of scientific works employing sequencing data has exponentially increased.
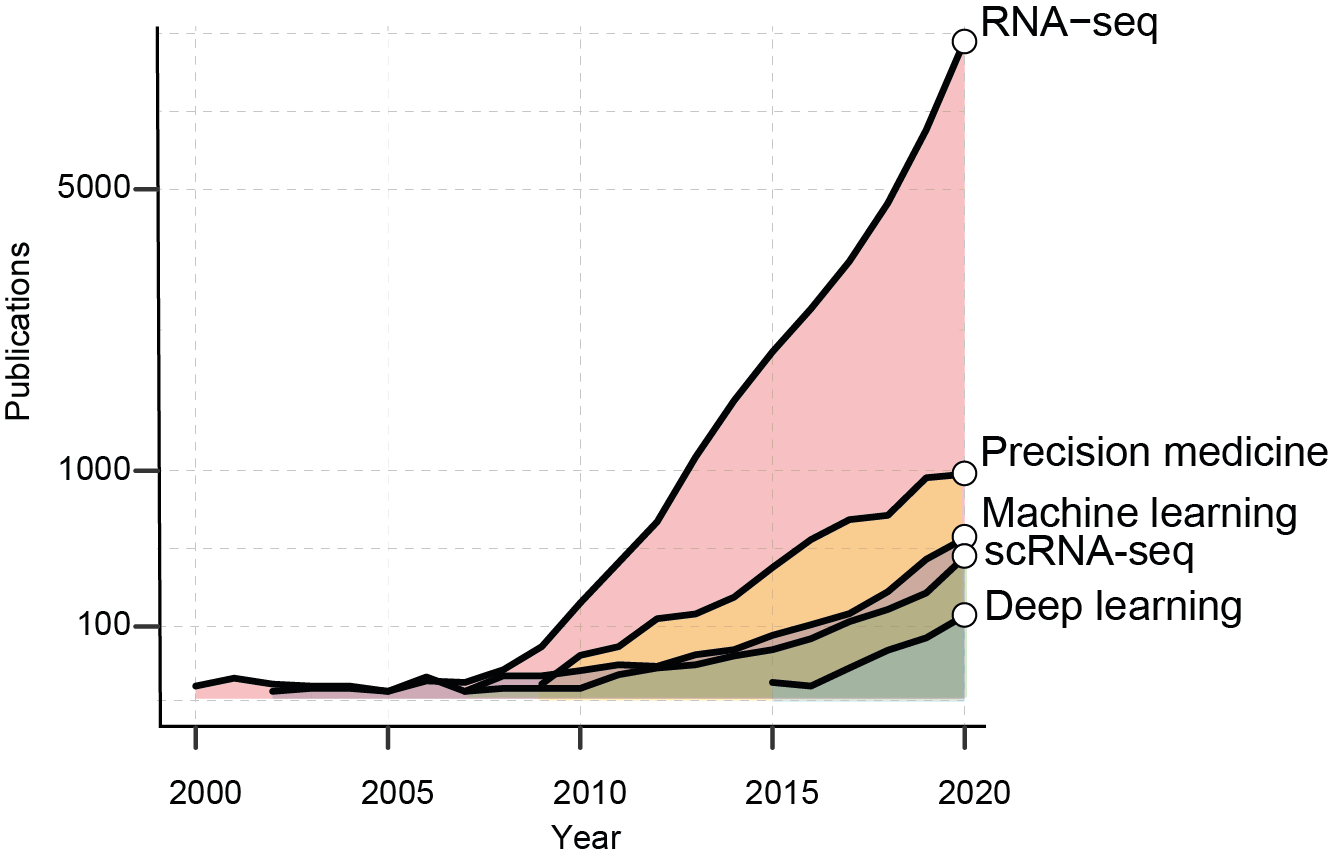
The growth of RNA-seq based studies. The graph shows the number of PubMed publications per years containing the reported keywords.
Machine and Deep Learning can leverage the heterogeneity of transcriptomic Big Data to achieve consistent predictions without the need of modeling the system of interest. These algorithms perform tasks that normally require human intelligence. While ML algorithms still need human guidance to improve their predictions, DL methods can autonomously determine the accuracy of a prediction.
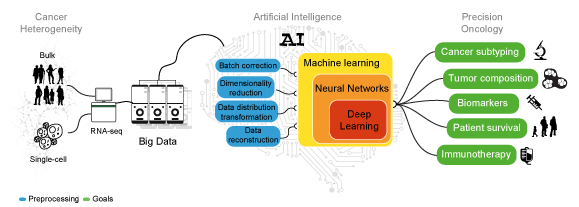
Artificial human intelligence. Sketch representing the analyses needed to decipher cancer heterogeneity and achieve an effective precision oncology.
Ourreview on Artificial Intelligence is out on International Journal of Molecular Sciences! ...We also have an R-based tutorial for AI development that is available here